
生长与连接模式都需要种子结构作为设计的起点。虽然LigBuilderv2.0提供了从已知抑制剂中提取种子结构的功能,但用户仍然需要认真检查这些种子结构。此外,用户也可以创建自己的种子结构,使得计算结果更符合用户的需求。我们先介绍一下如何人工的构建种子结构。
因为种子结构是所有设计结果的母体,选择合适的种子结构非常重要。种子结构最好能够体现出理想的设计目标的一些特性,例如,与受体发生强相互作用,或者是一个好的骨架,或者对合成来说是很好的起点。选择合理的种子结构有很多种方法,例如,可以从晶体复合物中的配体入手,通常配体中会有部分基团与受体有关键的相互作用,可以提取这些基团作为种子结构。如果用户已经通过其他手段获取了先导化合物,可以适当修剪之并对接进结合位点作为种子结构。也可以直接对接一些小的基团,例如氨,甲醛,苯等等进入结合位点作为种子结构。种子结构可以是很多个重原子组成,但也可以仅有一个重原子,例如甲烷。
种子结构以Mol2格式存储。需要注意的是种子结构中所有的化学价都必须是填满的,并且所有的氢原子都必须添加上。因为在格式转换时原子和键信息可能会丢失,所以需要仔细检查种子结构的原子类型的键类型是否正确。(我们建议用户使用“I-interpret”或者“openbabel”转换来自于PDB里的文件)。通过将"H"的原子类型改为"H.spc",用户可以在种子结构上将这些氢原子标记为 "growing sites(生长位点)"。用户可以在分子建模软件里编辑原子类型或者直接手动修改Mol2文件。BUILD模块会将所有被标记为"H.spc"的位点看做可以添加新片段的位置。特别是对于连接模式来说,把尽量多的氢原子标记为生长位点可以增加连接成功的概率。请注意:为BUILD模块准备的每个种子结构都应该是一个完整的化学分子,并且这些种子结构的坐标都应该位于结合位点中合适的位置。LigBuilder v2.0仅仅对种子结构进行很微弱的扰动,并不包括对接的步骤。
当用户第一次使用LigBuilder v2.0时,这些概念可能会难以理解。可以参考种子文件的例子"seed.mol2",该文件在"LigBuilderV2/example/receptor"目录下.
此外,我们提供了更为方便的种子提取器,用户可以通过“Extract”功能来从已知抑制剂中提取合适的种子结构。与人工创建种子结构一样,用于提取的目标抑制剂需要事先放置在结合位点中合适的位置。也就是说,在从复合物结构中获取配体前用户需要将所有复合物叠合到目标受体结构上,或者是将所有分子对接到目标受体中。需要注意的是种子结构中所有的化学价都必须是填满的,并且所有的氢原子都必须添加上。因为在格式转换时原子和键信息可能会丢失,所以需要仔细检查种子结构的原子类型的键类型是否正确。(我们建议用户使用“I-interpret”或者“openbabel”转换来自于PDB里的文件)。种子提取器会根据提取的模式判定生长位点,用户也可以编辑输出的种子结构进行修改。
下面是运行种子提取器的例子,可以参考"extract.input"文件,在"LigBuilderV2/example"目录中:
cd LigBuilderV2/example
./build -Extract extract.input
因为种子提取器更适合于进行仿制设计,用户也可以在 "LigBuilderV2/default/usersettings.input"中的仿制设计章节中找到相关设定集。
最后,用户可以通过设置"ADD_HSPC"参数为"YES"来将所有的氢原子视为为生长位点。
近年来有报道了很多判断类药性的经验规则,最著名的方法被称作Lipinski规则(Adv. Drug. Delivery. Rev. 1997, 23, 3-25)。根据Lipinski规则,当分子符合如下特性时吸收与转运较好:
如果分子违背上面的规则,则很可能不是一个合适的先导化合物。在LigBuilder V2.0中,我们采用类似的参数来控制设计的分子的类药性。我们也希望用户自己定义这些化学规则来进行跟有针对性的设计。BUILD中包括如下关键词:
"MAXIMAL_MOLECULAR_WEIGHT": 最大分子量
"MINIMAL_MOLECULAR_WEIGHT": 最小分子量
"MAXIMAL_LOGP": 最大LogP
"MINIMAL_LOGP": 最小LogP
LigBuilder v2.0使用XLOGP v2.0 的算法计算LogP。关于该算法的更多细节,请参考: Wang, R.; Gao, Y.; Lai, L. "Calculating partition coefficient by atom-additive method", Perspectives in Drug Design and Discovery, 2000, 19, 47-66.
"MAXIMAL_HB_DONOR_ATOM":最大氢键给体原子数。在这里给体原子指任何连接至少一个氢原子的氮原子或氧原子,也就是-OHs or -NHs
"MINIMAL_HB_DONOR_ATOM": 最小氢键给体原子数
"MAXIMAL_HB_ACCEPTOR_ATOM":最大氢键受体原子数。在这里受体原子指任何含有至少一对孤对电子的氮原子或氧原子
"MINIMAL_HB_ACCEPTOR_ATOM": 最小氢键受体原子数
"MAXIMAL_PKD":与靶标蛋白的最大结合强度。以pKd单位表示。
"MINIMAL_PKD": 与靶标蛋白的最低结合强度。以pKd单位表示。
LigBuilder v2.0使用 SCORE v4.0 的算法计算结合强度。该算法继承自SCORE V2.0。关于Score2的细节,请参考: Score v2.0:Wang, R.; Liu, L.; Lai, L.; Tang, Y. "SCORE: A new empirical method for estimating the binding affinity of a protein-ligand complex", J.Mol.Model., 1998, 4, 379-394.
从实际出发,一个合适的类药规则往往取决于用户所设计的体系。但对大多数情况来说,可以将设置如下参数:分子量:160~500;LogP:-0.4~5.6;氢键给体:2~5;氢键受体:2~5;预测pKd:7~12
在介绍章节里,我们已经提到过,LigBuilder v2.0使用分子片段作为构建单元来产生分子。LigBuilder v2.0中默认的分子构建单元库存储在目录"fragment.mdb"中。该库包含将近180个片段,包括大多数常见的化学基团和环骨架结构。每个片段都存储为Mol2格式。所有的片段都已经经过能量优化。
该目录中有一个"INDEX"文件,是构建单元的列表。用户可以编辑该文件以决定是否使用某个构建单元。"INDEX"文件的格式如下:
1 frag_01.mol2 1.00
methane
2
frag_02.mol2 0.50 ethane
3
frag_03.mol2 0.00 propane
......
这里每一行代表一个片段。第一列是片段的ID,每个片段都必须有自己独立的ID,不能相同。第二列式对应Mol2文件的名字。第三列代表改片段的"使用概率"。第四列是片段的名称。
需要注意的是"acceptability(使用概率)"这一列。该列的取值范围为0到1之间。如果设为1.00,则在构建新分子过程中,该片段会被100%的接受。如果设为0.00,则不会使用该片段构建分子。那该值在0到1之间表示什么呢?事实上,每次LigBuilder v2.0提取某一构建单元时,程序会产生一个0到1间的随机数,并与其使用概率进行比较。如果该随机数小于"使用概率",这个片段就被提取去构建分子,否则跳过该片段。 因此可以通过这个值来控制某个片段在设计中的使用概率。
虽然默认的构建单元库包含多种片段,但根据我们的经验,大多数情况下并不是所有的片段都需要被使用。换句话说,请仅使用必须的片段来构建分子。则可以节省计算时间并且使得计算结果更为合理。用户可以在INDEX文件中奖不想要的片段的使用概率改为0来屏蔽这些片段。
当然,我们并不限制用户必须使用默认的构建单元。用户可以自行添加新的片段。这个过程如下:(1)画出片段并保存为Mol2格式;(2)将此文件放入目录"fragment.mdb";(3)在"INDEX"文件中添加新的一行来定义该片段。进一步,用户还可以定义自己的构建单元库,这对于设计特定的组合化学库非常有意义。想要构建新的单元库,只需要将所有的片段收集到一个目录中,并在该目录下创建对应的INDEX文件,然后修改"default.input"中"Fragment Libraries"章节里的默认构建单元的目录。
在构建用户自己的片段时,请注意所有的片段都必须是完整、独立的分子,所有的化学价都必须是填满的,并且所有的氢原子都必须添加上。因为在格式转换时原子和键信息可能会丢失,所以需要仔细检查种子结构的原子类型的键类型是否正确。所有片段都需要进行能量优化,因为LigBuilder v2.0不进行这一过程。一般来说,刚性片段更为理想。如果片段包括可旋转键或者多种构象,请使用"典型"构象作为该片段的代表。当然,如果必要,也可以将多个构象存为不同的Mol2文件并标记为不同的片段。
需要注意的是每个片段都必须有至少一个生长位点。可以通过将对应的氢原子的原子类型从"H"改为"H.spc"来标记其为生长位点。当然,最简单的方法是设置所有的氢原子都是生长位点。用户也可以选择仅指定部分的氢原子为生长位点,通过这种方法可以根据用户的合成经验来控制分子设计过程。
此外,LigBuilder v2.0提供两个功能来自动构建片段库。"ExtLibrary"功能用于构建刚性片段库,"RotLibrary"功能用于构建多构象片段库。使用这些功能都会根据片段的出现频率来挑选片段,因此需要提供足够大的分子库来构建。
这儿还要说一下另外一个特殊功能。事实上构建单元库中的每个片段,并不能单纯的视为"一个"片段,而应该视为一个模板。在构建分子时,LigBuilder v2.0会通过对分子结构进行突变来提升结合能力。具有相同杂化类型的碳、氮、氧原子可以互相突变。例如,片段库中的"苯"除了表示它自身之外,还表示其他的6元芳香杂环。该策略使得用户不必过多的枚举所有的片段,仅需要提供模板即可。
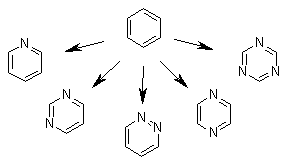
构建单元中的每一个片段实际上都是个模板
我们非常期望以及感谢用户发展新的构建单元并分享给其他的LigBuilder v2.0用户。您可以将您构建的构建单元发给我们,我们会将其添加到LigBuilder v2.0中并在网上更新。
构建分子时,LigBuilder v2.0通过一些内置的规则来屏蔽"不合理结构",例如-O-O,-N-O-,-N-N-等等,但这些规则可能还不足以构建合理的分子。因此,LigBuilder v2.0提供了外置的,可以由用户控制的片段库作为补充。我们称其为 "forbidden substructure library(禁止子结构库)"。在构建新的分子时,程序会检查每个分子是否包含禁止子结构库中的片段。如果有,则排除掉该分子。
配置文件中的"APPLY_FORBIDDEN_STRUCTURE_CHECK"参数用于控制是否启用禁止结构检查,可以是"YES" 或 "NO".
LigBuilder v2.0提供了一个默认的禁止子结构库。该库保存在"forbidden.mdb"目录中,每个结构都是Mol2格式。同样的,该目录下也有一个"INDEX"文件,列出库中所有的分子。INDEX文件的格式与"fragment.mdb"中的相同。用户可以编辑该文件来确定是否检查某个片段。请注意这里的第三列,也就是使用概率设定为1.00时,说明会检查该结构,当发现包含该子结构的分子时该分子会被排除。反过来,如果使用概率为0.00时,说明不检查该结构。因此,在这里设定除了0或1的值都没有意义。
目前默认的禁止子结构库仅有二十来个结构,用户有充分的发挥余地。我们希望用户自己添加新的禁止结构来完善这个库。操作方式如下:首先构建分子然后存为Mol2格式,然后将其置于目录下并在"INDEX"文件中添加对应的行。这个过程和添加结构单元库的方式相同。当然,这里不需要指定生长位点。
我们非常期望以及感谢用户定义新的禁止结构并分享给其他的LigBuilder v2.0用户。您可以将您构建的构建单元发给我们,我们会将其添加到LigBuilder v2.0中并在网上更新。
我们实验室也在做一些毒性和结构之间关系的研究,在考察毒性和分子子结构之间相关性后我们找到了将近80个具有毒性倾向的化学结构。关于这个工作的详细信息,请参考:Wang, J.; Lai, L.; Tang, Y. "Structural features of toxic chemical for specific toxicity", J.Chem.Inf.Comput.Sci., 1999, 39, 1173-1189.
我们将这些结构收集到"toxic substructure library(毒性子结构库)"中。该库保存在 "toxicity.mdb"目录里。配置文件中的"APPLY_TOXIC_STRUCTURE_CHECK"参数可以控制是否使用该库进行过滤,可以使"YES"或"NO"。如果使用该库,所有包含库中列出的子结构的分子都会被排除。
当然,用户也可以自己编辑该库。库中每个结构都是Mol2格式。同样,目录下也有一个"INDEX"文件列出了库中所包含的分子。INDEX文件的格式与"fragment.mdb"和"forbidden.mdb"中的相同。可以通过该文件控制是否在毒性检查时使用某一个片段。请注意这里的第三列,也就是使用概率设定为1.00时,说明会检查该结构,当发现包含该子结构的分子时该分子会被排除。反过来,如果使用概率为0.00时,说明不检查该结构。因此,在这里设定除了0或1的值都没有意义。
用户也可以自己添加新的结构。操作方式如下:首先构建分子然后存为Mol2格式,然后将其置于目录下并在"INDEX"文件中添加对应的行。事实上,这里不仅仅限制为毒性片段,任何可能影响设计结果合理性的结构,例如稳定性等都可以添加进去。用户可以将认为不适合在设计结果中出现的结构都加入这个库或者禁止库。
我们非常期望以及感谢用户定义新的毒性结构并分享给其他的LigBuilder v2.0用户。您可以将您构建的构建单元发给我们,我们会将其添加到LigBuilder v2.0中并在网上更新。
BUILD在产生每一代的设计结果后,都会将当前的种子库保存在由"SEED_RECORD_FILE"关键词定义的文件中,该文件是MOL2格式的。如果想恢复中断的任务,可以简单的用"Continue"或者 "DContinue"模式再次执行任务,程序会调用已经生成的种子库,继续运行。
Binding site(结合位点):
受体是整个设计过程的中心,在开始设计前,用户需要仔细的检查目标受体的情况。关键的结构信息,特别是结合位点附近的突变、诱导契合效应、柔性、构象变化、金属离子、不确定的原子(原子占据率不为1的时候),都需要仔细考虑。大多数情况,分辨率较高、与一个"较大的"配体结合的复合物结构更适合用于设计。
然后使用CAVITY模块可以得到对受体结合位点的探测结果。请用户确认哪一个探测结果是目标结合位点。如果探测结果不理想,用户可以更改CAVITY的探测参数然后再次探测,直到探测结果能够很好地描述所需要的结合位点。the desired design area accurately.
CAVITY会预测结合位点最强的结合能力(pKd)。如果该值小于7.0(100nM),则需要调整探测参数来提高该数值,或者,说明这个结合位点可能并不是合适的药物设计位点。
Molecule diversity and synthesis accessibility(分子多样性和可合成性):
通常来说,分子的多样性高是不利的,因为这样会使得可合成性下降,同时因为化学空间非常巨大,我们有很大可能找到复杂度低但活性高的分子,也不需要很高的多样性。但是,有的时候我们可以通过增加多样性来发现新的骨架结构以及进一步提升活性,所以用户可以根据自己的目标来决定设计的多样性。
因为我们已经事先对合成模块的分析速度和精度做了较好的平衡处理,建议用户不要随意修改合成模块的参数。用户可以直接调用预置的参数集来调整合成的参数。
Binding affinity(结合强度):
默认的输出pKd下限是7.0(100nM),用户可以根据CAVITY对结合位点最高pKd的预测结果在 "build.input"中修改"$GOAL_PKD$"参数。
Bioactivity(活性):
因为合成新化合物难度远高于购买已有化合物,全新药物设计必须达到远低于虚拟筛选的假阳性率。 LigBuilder v2.0提供了一个"recommender(挑选器)",通过统计的算法来提升富集率。用于挑选的基数越大,挑选的精度越高。所以对一个体系设计非常大量的分子很重要,设计得越多越好。
先导化合物可以被用于进一步的设计或者通过实验进行衍生。LigBuilder v2.0可以设计低分子量、高原子经济性的先导化合物。
用户可以通过调用"LigBuilderV2/default/usersettings.input"中的Lead Discovery章节中的先导设计参数集,启用先导设计策略。先导设计模式会限制设计分子量在160~300 Da间,并且扩大LogP的范围到-2.4~7.6之间。这里,推荐用户将"$GOAL_PKD$"设为6.0或以下。请注意:最好在探索模式中使用先导设计策略。
启用先导设计策略后,用户仍然使用正常探索模式一样的方式运行BUILD。
BUILD可以将先导化合物或者已有的分子作为起始结构进行优化生长。这些起始结构被用作生长模式或连接模式的种子(DESIGN_MODE 设为1或2)。用户必须确保这些分子坐标都已经事先放置在结合位点中合适的位置,否则用户需要先将他们对接进结合位点。需要注意的是种子结构中所有的化学价都必须是填满的,并且所有的氢原子都必须添加上。因为在格式转换时原子和键信息可能会丢失,所以需要仔细检查种子结构的原子类型的键类型是否正确。(我们建议用户使用"I-interpret"或者"openbabel"转换来自于PDB里的文件)。用户可以根据"如何构建种子结构"章节设定生长位点,或者简单的通过设置"ADD_HSPC"为"YES"将所有氢原子都视为生长位点。然后,将所有需要用到的种子结构的路径,放入一个列表文件,并将列表文件路径写在"SEED_LIGAND_LIST"参数中。
最后,用户可以如运行普通模式一样,运行BUILD来进行生长模式或者连接模式的计算。
因为可以利用已知抑制剂、活性天然产物、甚至是缺乏发展潜力的抑制剂的信息,相较于全新设计,仿制设计有许多的优势。我们知道,具有靶标蛋白活性的分子往往具备重要的相互作用模式或者是关键作用基团,因此来自于活性分子的片段可以视为潜在的结合锚定片段。BUILD可以从已知分子中提取这些锚定片段并尝试将其重新组合成新的分子。显而易见,使用这种策略设计的分子,应当更为可靠。BUILD所采用的提取策略倾向于提取关键的相互作用而不是骨架结构。BUILD将会通过形成新的结构来连接这些相互作用基团,因此能够保证骨架结构的新颖性,可以有效地减少与专利的冲突。当然,用户需要提供足够的具有不同结合模式的抑制剂。
在从已知结构提取种子之后(请参考“如何构建种子结构”章节),我们建议用户对提取的结构进行人工的核查以去除掉不合理或者不满意的结构。用户也可以人工添加关键的片段。
用户可以通过在"LigBuilderV2/default/usersettings.input"中的mimic design章节调用仿制设计参数集来启用仿制设计功能。请注意:最好在连接模式中使用仿制策略(如果种子结构大于5个),或者在生长模式(如果种子结构小于等于5个)。对连接模式来说,如果提取的种子结构非常充足(种子结构数大于20),用户可以增加"MINIMAL_LINKING_NUMBER"参数的值。默认值为2,也就是说,BUILD会设计包含2个种子片段的分子。
最后,用户可以如运行普通模式一样,运行BUILD来进行生长模式或者连接模式的计算。
[Content] [Introduction] [Download] [Install] [Overview] [CAVITY] [BUILD] [Skills] [FAQs]
(These web pages are edited by Yaxia Yuan. Latest update: Jan., 2012)